My research interest lies in the application of deep learning to de novo drug design and discovery. I am currently exploring the implementation of deep generative models, particularly variational autoencoders (VAEs), in this field. My bachelor’s thesis is a testament to this, as it focuses on the use of VAE models and Bayesian optimization, in conjunction with traditional drug discovery tools such as QSAR and Molecular docking, to identify new potential drug candidates with real-world applications. I seek to contribute to the advancement of this domain through novel approaches and applications.
Discovery of Vascular Endothelial Growth Factor Receptor 2 Inhibitors Employing Junction Tree Variational Autoencoder with Bayesian Optimization and Gradient Ascent
Gia-Bao Truong, Thanh-An Pham, Van-Thinh To, Hoang-Son Lai Le, Phuoc-Chung Van Nguyen, The-Chuong Trinh, Tieu-Long Phan, and Tuyen Ngoc Truong
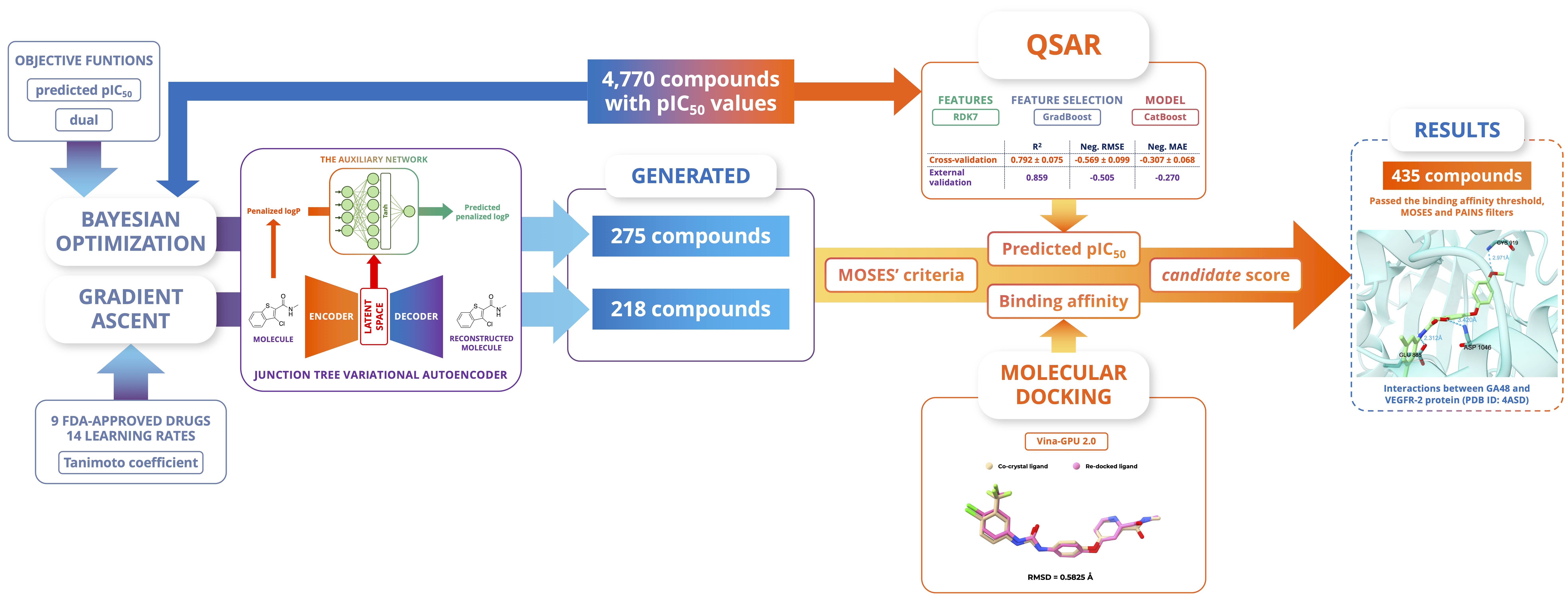
In the development of anticancer medications, vascular endothelial growth factor receptor 2 (VEGFR-2), which belongs to the protein tyrosine kinase family, emerges as one of the most significant targets of interest. The ongoing Food and Drug Administration (FDA) approval of novel therapeutic medicines toward VEGFR-2 emphasizes the urgent need to discover sophisticated molecular structures that are capable of reliably limiting VEGFR-2 activity. Recognizing the huge potential of deep-learning-based molecular model advancements, we focused our study on exploring the chemical space to find small molecules potentially inhibiting VEGFR-2. To achieve this goal, we utilized the junction tree variational autoencoder in combination with two optimization approaches on the latent space: the local Bayesian optimization on the initial data set and the gradient ascent on nine FDA-approved drugs targeting VEGFR-2. The optimization results yielded a set of 493 uncharted small molecules. Quantitative structure–activity relationship (QSAR) models and molecular docking were used to assess the generated molecules for their inhibitory potential using their predicted pIC50 and binding affinity. The QSAR model constructed on RDK7 fingerprints using the CatBoost algorithm achieved remarkable coefficients of determination (R2) of 0.792 ± 0.075 and 0.859 with respect to internal and external validation. Molecular docking was implemented using the 4ASD complex with optimistic retrospective control results (the ROC-AUC value was 0.710 and the binding activity threshold was −7.90 kcal/mol). Newly generated molecules possessing acceptable results corresponding to both assessments were shortlisted and checked for interactions with the protein at the binding site on important residues, including Cys919, Asp1046, and Glu885.
@article{vegfr2,title={Discovery of Vascular Endothelial Growth Factor Receptor 2 Inhibitors Employing Junction Tree Variational Autoencoder with Bayesian Optimization and Gradient Ascent},author={Truong, Gia-Bao and Pham, Thanh-An and To, Van-Thinh and Lai Le, Hoang-Son and Van Nguyen, Phuoc-Chung and Trinh, The-Chuong and Phan, Tieu-Long and Truong, Tuyen Ngoc},keywords={Vascular endothelial growth factor receptor 2, junction tree variational autoencoder, bayesian optimization, gradient ascent},doi={10.1021/acsomega.4c07689},url={https://pubs.acs.org/doi/10.1021/acsomega.4c07689},journal={ACS Omega},year={2024},}